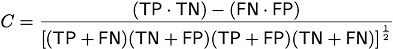 ,
,| RNA Home | Aalberts' Lab |
|---|
,A common bioinformatics problem is how to determine whether a local primary sequence X is more like examples in real or decoy training data sets.
Many methods (Weight Matrix Method, Weight Array Method, Markov Models) have been developed to address this problem. A WMM-type approach uses the probabilities of inidividual letters [p(e)>p(t)>...>p(z)] at different positions. Correlations [e.g., qu or th] are included in more sophisticated models.
Our Primary Sequence Ranking methods take a complementary approach, more akin to referring to a dictionary to see whether the word is known. And if the real and decoy dictionaries are small (abridged), we also offer several approaches to enhance the lexicon by making substitution mutations. In the strange world of bioinformatics, local sequence X can appear in both real and decoy dictionaries. (In fact, in our paper we show that in the case of pre-mRNA donor splice signals, 96% of real 7 letter sequences also appear as decoys elsewhere where they are not spliced.)
The PSR approach is very simple. We rank order sequences by the likelihood X is true or '+':
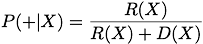
To accomodate finite training data, we also provide ways of enhancing the data by including pseudocounts from nearest-neighbor X' and next-nearest-neighbor X'' sequences (one and two substition mutations from X) .
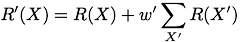
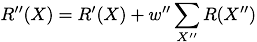
Quantifying Optimal Accuracy of Local Primary Sequence Bioinformatics Methods, Daniel P. Aalberts, Eric G. Daub '04, and Jesse W. Dill '04, Bioinformatics 21, 3347-3351 (2005).